Nous
avons tous assisté à ces tours de « magie » où
deux cordes formant deux anneaux joints se séparent subitement entre
les mains expertes qui les manipulent. Depuis longtemps, les biologistes
ont observé que des enzymes, les topoisomérases, réalisent
ce numéro d’illusionniste sur les molécules d’ADN
de nos chromosomes. L’enjeu est de taille. Lorsqu’au sein de
nos cellules la double hélice « mère » est recopiée,
les deux molécules « filles » qui en résultent
s’entortillent l’une autour de l’autre dans un formidable
enchevêtrement de nœuds. Sans ces topoisomérases, la séparation
de ces deux molécules « filles » est impossible et la
cellule meurt au lieu de se diviser normalement. Pour enlever ces nœuds,
les topoisomérases coupent une molécule d’ADN en deux
tout en maintenant ses deux extrémités, puis font passer une
autre molécule d’ADN à travers cette brèche.
Finalement, elles recollent parfaitement la molécule coupée.
Les hélicases sont elles capables de séparer les deux brins
de la double hélice. Cette opération est essentielle pour
de nombreuses étapes de la vie cellulaire, un défaut de fonctionnement
de ces enzymes entraîne des conséquences catastrophiques pour
la cellule. En utilisant une bille magnétique micrométrique
attachée à deux molécules d’ADN, il est maintenant
possible d’observer ces tours de passe-passe moléculaires en
temps réel et ainsi de mieux comprendre leur fonctionnement.
Le contexte biologique :
Il y a 50 ans, Crick et Watson décrivaient la structure de la double hélice et faisaient remarquer que l’existence des deux brins complémentaires permettait de proposer un mécanisme de réplication de la molécule dans lequel il suffit de séparer les deux brins et de recopier chacun en formant son complémentaire pour obtenir enfin deux molécules d’ADN filles rigoureusement identiques à la molécule parente. Cette intuition devint une réalité dans les années 1970-1980 lorsque les enzymes chargées du recopiage, les polymérases, ainsi que celles chargées de séparer le brins complémentaires, les hélicases, furent identifiées puis isolées .
La structure de la double hélice
implique que les deux brins d’ADN complémentaires s’entortillent
l’un autour de l’autre. Par ailleurs, les molécules
d’ADN sont très longues (~10 cm pour un chromosome) et l’on
compte un tour d’hélice tous les 3,4 nanomètres. En
conséquence, le nombre de tours impliqué est gigantesque.
Or, le bon fonctionnement du mécanisme de duplication de l’ADN
implique que tous les tours, jusqu’au dernier, soient débobinés
afin de pouvoir séparer les deux molécules filles. Afin
d’accommoder cette contrainte majeure et très délicate,
on imaginait simplement que les molécules tournaient au niveau
de leur extrémités. Cette hypothèse simple ne peut
malheureusement être retenue, puisque dans le cas des bactéries,
le génome se trouve sous la forme d’une molécule circulaire
d’ADN, c’est-à-dire une double hélice refermée
sur elle-même. En conséquence, les deux brins de la double-hélice
sont topologiquement inséparables. Dix ans après la découverte
de Crick et Watson, ce problème a même poussé certains
à dire que la double hélice n’était probablement
pas la bonne structure !
C’est James Wang qui dans les années 70 apporta la solution à l’énigme [1]. Il découvrit une nouvelle classe d’enzyme, les topoisomérases, qui sont capables de changer la topologie de la molécule d’ADN en effectuant une coupure temporaire dans la molécule pour y faire passer soit un brin, soit les deux brins de la double hélice. Ce faisant, ces enzymes permettent de relâcher les contraintes de torsion sur une molécule ou de désenchevêtrer deux molécules entortillées.
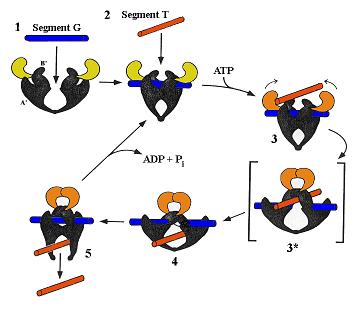
Figure 1 Schéma simplifié décrivant le mécanisme moléculaire d'action des topoisomérases de type II. L’enzyme correspond à l’objet symétrique noir avec une partie mobile jaune ou noire. Dans la configuration initiale (1) l’enzyme s’ouvre pour accueillir une première molécule d’ADN (segment G bleu) puis une seconde (segment T rouge). Une fois les deux molécules en place, l’enzyme accroche deux molécules d’ATP et coupe le segment G (gate : porte en anglais) qui laisse alors passer le segment T (transporté) au travers de cette brèche. La topoisomérase recolle alors la molécule bleue avant de relâcher les deux molécules. Le bilan global de cette réaction enzymatique est ainsi d’inverser le sens du croisement des molécules bleue et rouge. (Ce schéma proposé par J. Wang est un peu simplifié par rapport au modèle actuellement accepté).
La découverte des topoisomérases a permis de résoudre le problème des nœuds dans les molécules d’ADN. Cependant, leur fonctionnement a soulevé d’autres questions : comment des enzymes mesurant quelques nanomètres peuvent-elles relâcher jusqu’au dernier tour d’entortillement des molécules qui s’étendent sur des distances jusqu’à un million de fois plus grandes ? Comment ces enzymes déterminent de quel côté de la brèche il faut transférer une molécule afin de défaire un nœud et non pas, au contraire, en créer de nouveaux ? Les réponses à ces questions nous manquent encore à l’heure actuelle. Certains de nos résultats suggèrent que ces enzymes reconnaissent l’angle formé par les molécules lors de leur croisement. En particulier, un type de topoisomérase que l’on trouve chez les bactéries se comporte très différemment selon que cet angle est positif et négatif. Cependant, les vérifications expérimentales de ces hypothèses sont particulièrement délicates à réaliser dans des expériences de biologie classique, c’est-à-dire faites en tube à essai. En effet, dans ce contexte, comme les molécules d’ADN sont soumises à l’agitation thermique, l’angle qu’elles adoptent lors de leur croisement est largement aléatoire.
Néanmoins, depuis quelques années, des expériences peuvent être réalisées à l’échelle d’une seule molécule. Ces expériences permettent de contrôler les paramètres physiques d’une molécule et d’imposer, comme nous le décrivons ci-dessous, l’angle de croisement entre deux molécules.
La micromanipulation par pinces magnétiques :
Ce sont les groupes de S. Chu et C. Bustamante qui ont réalisé les premières expériences de micromanipulation de molécules uniques [2]. Celles-ci se font à l’aide d’un microscope optique et dans le milieu naturel de la molécule d’ADN, c’est-à-dire de l’eau avec un peu de sel. Dans ces conditions, l’observation directe de la molécule est impossible. Par contre, en utilisant un « scotch moléculaire » il est assez facile d’attacher une bille de quelques microns à une extrémité de la molécule. Ceci permet d’observer la bille simplement au microscope pour en déduire la position de l’extrémité de la molécule d’ADN qui lui est attachée. Mieux, on peut attacher de façon analogue la seconde extrémité de la molécule à la paroi du récipient. En exerçant une force sur la bille, on peut ainsi étirer la molécule d’ADN. Le « scotch moléculaire » est un couple de molécules de type « clef/serrure » : la clef est une petite molécule (de la biotine par exemple) qui possède une affinité très importante pour une molécule plus grosse qui en épouse la forme (de la streptavidine dans le cas de la biotine). La molécule d’ADN est préparée en attachant chimiquement la biotine à une extrémité. Les billes magnétiques sont recouvertes de streptavidine. En plaçant les molécules d’ADN ainsi préparées en présence des billes en solution, celles-ci se couplent spontanément aux billes de façon quasi-irréversible. L’accrochage de la seconde extrémité de la molécule se fait par un deuxième jeu clef/serrure (digoxigénine/anti-digoxigenine). Comme l’accrochage des billes aux molécules d’ADN se fait par diffusion, rien n’empêche deux ou plusieurs molécules d’ADN de relier la bille à la surface du récipient. En pratique, nous ajustons la concentration relative des molécules aux billes pour que la plupart de celles-ci soient reliées par une seule molécule d’ADN. Cependant, statistiquement un petit nombre de billes se trouvent être attachées par deux molécules d’ADN, cette configuration va nous permettre de croiser à volonté deux molécules.
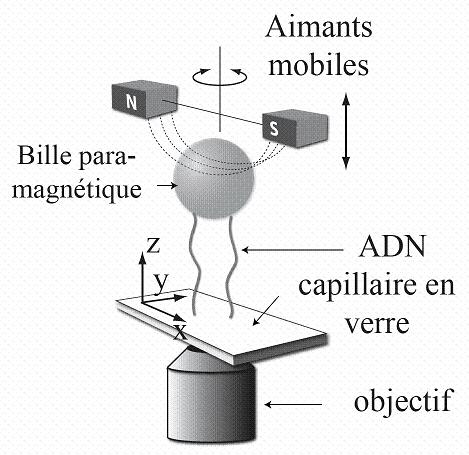
Figure 2 Principe de l'expérience de micromanipulation.
L’observation se fait à l’aide d’un microscope
optique placé sous l’échantillon. Les molécules
d’ADN sont invisibles. En revanche, les billes matérialisent
leurs extrémités. L’échantillon est constitué
par un tube de verre de section parallélépipédique
dans lequel nous avons introduit des molécules d’ADN qui
s’accrochent à la paroi du tube de verre et à des
micro-billes magnétiques. Des aimants (dont les dimensions ne sont
pas respectées sur ce schéma) sont placés au dessus
de l’échantillon, ils exercent une force de traction verticale
d’autant plus grande que les aimants sont proches. En faisant tourner
les aimants autour de l’axe vertical, nous faisons tourner les billes
sur elles-mêmes. Dans l’échantillon, toutes les billes
ne sont pas forcément attachées par deux molécules
mais celles qui le sont présentent un changement d’extension
décelable lorsque les deux molécules sont amenées
à se croiser (voir Fig. 3). Il est ainsi facile de choisir les
billes attachées par deux molécules.
Au cours de leur mouvement brownien, les molécules d’eau bousculent l’ADN dans tous les sens et tendent à lui faire adopter la forme d’une pelote fluctuante. Il faut donc appliquer une force pour étirer la molécule ; ceci se fait en agissant sur la bille micrométrique qui localise une extrémité de la molécule . Il existe plusieurs moyens pour appliquer cette force. D’abord, les pinces optiques, qui utilisent un faisceau laser convergeant qui attire la bille près de son point de focalisation. Ensuite, les pinces magnétiques, basées sur l’utilisation d’aimants qui attirent la bille contenant un matériau magnétique. Cette seconde méthode permet également de faire tourner la bille simplement en faisant tourner les aimants.
Dans le cas des pinces magnétiques, pour une position donnée des aimants par rapport à la bille, la force appliquée est constante dans le domaine que peut explorer la bille. Pour déterminer cette force, il suffit de mesurer l’ampleur du mouvement brownien de la bille. Pour les faibles forces ces mouvements sont importants ; plus on rapproche les aimants, plus la force de traction augmente et plus l’ampleur du mouvement brownien diminue. La bille attachée à la molécule d’ADN sous l’action des aimants se comporte comme un pendule inversé. En appliquant le théorème d’équipartition de l’énergie, on montre que F = l.kBT/<x2> [3] (où l est l’extension de la molécule, <x2> est l’amplitude quadratique moyenne du mouvement brownien, kB la constante de Boltzmann et T la température absolue). La force typique qu’il faut appliquer pour étirer une molécule d’ADN est de l’ordre de 1 pN (10-12 N), mille fois plus faible que la force de rupture de l’ADN (~1000 pN).
Pour mesurer le mouvement brownien,
nous avons écrit un programme de traitement d’images vidéo
qui suit la position horizontale de la bille en temps réel, avec
une précision de quelques nanomètres. En analysant les motifs
de diffraction de l’image de la bille obtenue en éclairage
parallèle, il est également possible d’obtenir la
position verticale de la bille avec une précision comparable. Cette
mesure nous permet ainsi de déterminer la distance séparant
la bille de la paroi du récipient.
Le vrillage d’une balançoire
Il est assez facile de sélectionner les billes ancrées à la paroi par deux molécules : ces molécules sont typiquement séparées par une distance comparable au rayon de la bille. En faisant tourner celle-ci d’un demi-tour dans un sens ou dans l’autre, les deux molécules sont amenées à se croiser. Si la longueur des molécules est comparable au diamètre de la bille, il se produit alors un raccourcissement notable de la distance séparant la bille à la paroi, comme on peut l’observer en faisant vriller une balançoire autour de son axe. Cette variation rapide d’extension sur un tour n’est visible que sur des billes accrochées par deux molécules. Ces billes nous fournissent un moyen simple de croiser deux molécules avec un angle donné que nous évaluons en mesurant le raccourcissement provoqué par un demi-tour comparé à la longueur des molécules d’ADN dans leur configuration parallèle. Evidemment, pour une longueur donnée de la molécule d’ADN, cet angle dépend de la distance séparant les deux molécules que nous ne contrôlons pas. Cependant en allant à la pêche aux billes, on peut réaliser un échantillonnage de différents angles de croisement s’étalant de 50° à plus de 100° [4].
Tours de passe-passe moléculaire.
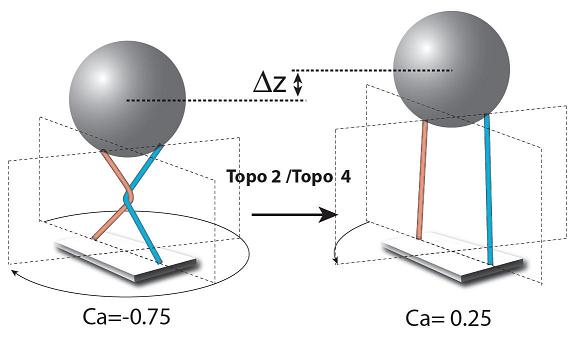
Figure 3 Principe d’action de la topoisomérase sur le croisement de deux molécules d'ADN. En faisant faire un tour à la bille avec les aimants, nous pouvons passer d’une situation où les molécules ne présentent pas de croisement (droite) à celle où elles se croisent (gauche). La topoisomérase reconnaît alors le croisement, et en opérant l’action décrite dans la figure 1, l’enzyme dénoue le croisement et ramène les deux molécules dans la situation sans croisement (droite). Dés lors, l’enzyme ne peut plus agir. Le changement de hauteur entre les deux configurations permet de déterminer le moment où l’action de l’enzyme s’effectue
Si nous ajoutons maintenant des topoisomérases dans la solution avec un peu d’ATP (la source d’énergie nécessaire à la plupart des opérations enzymataiques), il ne se passe rien si les deux molécules sont parallèles. Par contre, si nous créons un point de croisement en imprimant un tour à la bille, l’extension de la molécule diminue pour les raisons décrites ci-dessus. Alors, une topoisomérase va s’accrocher au croisement et le supprimer, permettant à l’extension de reprendre sa valeur maximale de départ.. On peut alors imprimer un nouveau tour à la bille générant un nouveau croisement que l’enzyme va s’empresser de dénouer, etc. Chaque événement correspond à un seul cycle enzymatique d’autant plus facile à détecter que le changement d’extension correspond à une fraction de micron comme on peut le voir sur la figure 4.
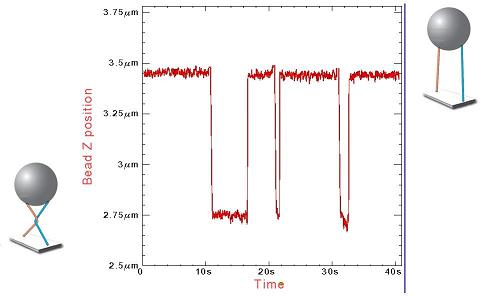
Figure 4 Signal expérimental permettant de voir
l’action de la topoisomérase : le graphique représente
la distance entre la bille et la paroi de verre. Lorsque celle-ci atteint
la valeur de 3.45 microns, les deux molécules sont parallèles.
Quand les molécules se croisent la distance se réduit à
2.75 microns. En présence de topoisomérases dans la solution
on n’observe aucun changement de longueur lorsque les molécules
sont parallèles. Par contre chaque fois que l’on fait faire
un tour aux aimants, dans un premier temps, la bille se rapproche de la
paroi. Au moment où l’enzyme dénoue le croisement,
on observe une remontée brutale de l’extension. Sur cet enregistrement,
nous avons répété l’opération trois
fois, à chaque fois l’enzyme a agit, cependant elle l’a
fait après un temps très variable.
Le temps mis par l’enzyme pour libérer le croisement après sa formation correspond au temps de diffusion de l’enzyme pour trouver le point de croisement et au temps de fixation sur ce croisement. Il dépend évidemment de la concentration d’enzyme; mais d’un cycle au suivant ce temps est une variable aléatoire présentant une distribution statistique de Poisson avec un temps caractéristique ?. Pour une concentration enzymatique de l’ordre du nano-molaire, ? est typiquement de quelques secondes. Si nous tournons la bille de plusieurs tours rapidement, après un temps d’attente, une enzyme enchaîne une série de cycles avec une cadence de 2 ou 3 à la seconde [5].
Les topoisomérases reconnaissent l’angle de croisement
des molécules.
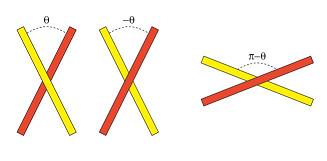
Figure 5 : Symétrie angulaire impliquée
dans le croisement de deux molécules d'ADN. La situation correspondant
à l'angle -? (au centre) est différente de la situation
θ (à gauche) par contre elle est équivalente à
π - θ (à droite). Notons que pour θ
= 90 les différentes configurations sont identiques.
Le point de croisement de deux molécules d’ADN formant un angle θ n’est pas le symétrique de la configuration correspondant à l’angle -θ. Par contre, les situations -θ et π - θ sont, elles, symétriques [6]. Dans notre expérience, en tournant la bille d’un demi-tour dans le sens des aiguilles d’une montre nous obtenons un angle de croisement θ en tournant dans l’autre sens, nous obtenons -θ. En employant la rotation des aimants pour génerer le substrat topologique (c’est à dire l’angle) voulu, il est aisé de mesurer le temps mis par la topoisomérase IV pour dénouer le croisement correspondant à θ et -θ. La valeur de l’angle θ est déterminée à partir du changement de hauteur de la bille et de la longueur des molécules.
Pour les petits
angles de croisement (correspondant à des billes dont la hauteur
change peu en passant de la configuration molécules parallèles
à molécules croisées), nous observons que le temps
d’action moyen de la topoisomérase IV est vingt fois plus
long pour la configuration à 50° que celle correspondant à
–50°. Pour les billes qui présentent une variation de
hauteur importante, l’angle de croisement approche 90° et peut
même dépasser cette valeur. Or comme nous l’avons expliqué
la situation de croisement à 90° est identique à celle
de croisement à -90°. Ainsi nos expériences montrent
que pour un angle de θ = 76° ou -76° les temps moyens
d’action ne diffèrent que de 10 %.
Puisque les molécules d’ADN sont animées de fluctuations
browniennes importantes, leur angle de croisement présente en fait
une distribution et la valeur de l’angle de croisement dont nous
avons parlé est en fait la valeur moyenne. La largeur de cette
distribution dépend de la force de traction appliquée aux
molécules. Si la force est très grande, les molécules
sont quasiment rectilignes et la distribution des angles est étroite
; aux faibles forces c’est l’inverse. Dans notre expérience,
nous observons bien que la sélectivité angulaire est renforcée
avec la force appliquée sur la bille.
La topoisomérase IV
est très habile lors de son tour de passe-passe moléculaire,
jamais nous ne l ‘avons prise en défaut de lâcher les
brins coupés avant de les recoller (la bille se retrouverait alors
accrochée par une seule molécule). Un tel accident serait
dramatique au sein de nos chromosomes : il conduirait à une cassure
double brin qui peut certes être réparée par des mécanismes
cellulaires adaptés mais avec un taux d’échec très
gênant.
L’ouverture de la molécule d’ADN comme une fermeture
éclair par une hélicase.
Lors de la réplication, les deux brins qui constitue la double hélice sont séparés par une enzyme, l’hélicase, qui se déplace en s’accrochant à l’un deux et en expulsant le second. Cette opération n’est pas très facile à observer dans un tube à essai sur une molécule un peu longue car sans le cortège d’enzymes qui accompagne l’hélicase dans la fourche de réplication au sein de la cellule, les deux brins séparés sont toujours complémentaires et ils s’associent de nouveau rapidement pour reformer la double hélice.
Le dispositif de pinces magnétique que nous venons de décrire pour les topoisomérases peut être facilement utilisé pour regarder les hélicases. Il suffit de remplacer les deux molécules d’ADN par une seule molécule simple brin dont la séquence est localement auto-complémentaire. Ceci entraîne le repliement de la molécule sur elle-même, formant ainsi une boucle à cheveux. La bille magnétique est attachée à une extrémité de cette boucle tandis que la seconde est attachée à la surface de verre comme on peut le voir sur la figure 6 :
Figure 6 : Gauche : une molécule d’ADN simple brin partiellement auto-complémentaire est ancrée à la surface du capillaire de verre. La bille magnétique qui lui est attachée permet d’ouvrir et de refermer mécaniquement la portion auto-appariée. Droite : Lorsque l’on maintient la force à une valeur inférieure au seuil d’ouverture de la molécule d’ADN, on peut observer l’activité d’une hélicase (ici l’hélicase réplicative du bactériophage T4). La molécule d’ADN passe la majorité du temps repliée (son extension vaut ici environ 290 nm), mais lorsqu’une hélicase se fixe et ouvre l’ADN, son extension varie de manière importante (pics).
Tant que la force de traction appliquée par la bille est faible,
la boucle à cheveux reste fermée, au delà de ~15pN
la double hélice s’ouvre spontanément. Cette ouverture
est facilement mesurée par l’allongement de la molécule.
Si maintenant nous appliquons une force inférieure à cette
force d’ouverture, la boucle reste fermée, elle peut cependant
s’ouvrir si une hélicase présente dans la solution
trouve cette molécule d’ADN. Cette fois l’ouverture
se fait progressivement en suivant le rythme de l’enzyme. Une fois
la boucle ouverte, l’hélicase se détache de la molécule
d’ADN et la boucle se reforme après quelques secondes. La
molécule est revenue à son état initial jusqu’à
ce qu’une nouvelle hélicase se décide à l’ouvrir.
On observe ainsi une série d’ouvertures séparées
par de longue phase d’attente comme on peut le voir sur la figure
6.
Conclusion
Nous venons de décrire une méthode expérimentale simple qui nous a permis de montrer l’existence d’un angle préférentiel d’action des topoisomérases sur les molécules d’ADN et d’observer l’action d’une hélicase. Cette méthode repose sur les techniques de micromanipulation de molécules uniques. C’est actuellement un sujet de recherche très actif et plusieurs groupes de recherche ont obtenu des résultats remarquables sur les moteurs moléculaires, les polymérases, les hélicases etc. Ces résultats viennent naturellement compléter ceux obtenus en tube à essai. Ils démontrent, s’il était nécessaire, que ces enzymes sont de magnifique machines capables de travailler avec une remarquable précision dans un environnement agité par le mouvement brownien. Les topoisomérases sont pour le moins des enzymes extraordinaires, et en bien des points elles surpassent ce que nous savons faire à l’échelle macroscopique. Ainsi il nous reste encore à comprendre comment ces machines de taille manométrique dénouent fidèlement des molécules mille fois plus grandes qu’elles.
Remerciements : Ces expériences n’auraient pas été possible sans l’aide de O. Saleh, H. Yokota, M. Duguet et le support financier de l’ENS, du CNRS, des universités Paris VI et VII, de la CEE et de l’ARC.
Références :
[1] Wang. JC. Interaction between DNA and an Escherichia coli protein omega. J Mol Biol. 1971 Feb 14;55(3):523-33.
[2] Smith SB, Finzi L, Bustamante C. Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads. Science. 1992 Nov 13;258(5085):1122-6.
[3] The elasticity of a single supercoiled DNA molecule TR. Strick, JF. Allemand, D. Bensimon, A. Bensimon, V. Croquette, Science (1996) 271-5257 p.1835
[4] Charvin G, Bensimon D, Croquette V. Single-molecule study of DNA unlinking by eukaryotic and prokaryotic type-II topoisomerases. Proc Natl Acad Sci U S A. 2003 Aug 19;100(17): 9820-5.
[5] Stone MD, Bryant Z, Crisona NJ, Smith SB, Vologodskii A, Bustamante C, Cozzarelli NR. Chirality sensing by Escherichia coli topoisomerase IV and the mechanism of type II topoisomerases. Proc Natl Acad Sci U S A. 2003 Jul 22; 100(15): 8654-9
[6] Timsit Y, Duplantier B, Jannink G, Sikorav JL. Symmetry and chirality in topoisomerase II-DNA crossover recognition. J Mol Biol. 1998 Dec 18;284(5):1289-99.